
mgr inż. Karolina Kosińska
Pracownik Katedry Biotechnologii i Biologii Komórki Wyższej Szkoły Informatyki i Zarządzania w Rzeszowie, absolwentka studiów na kierunku Biotechnologia na Politechnice Rzeszowskiej o specjalizacji Biochemia Stosowana (studia inżynierskie) oraz Biotechnologia Farmaceutyczna (studia magisterskie). Prywatnie sympatyk miasta Gdynia oraz polskiego morza, a także sympatyk biologiczno-chemicznych treści popularnonaukowych.
Choroba CWD manipuluje świadomością jeleni w USA – czy stanowi również zagrożenie dla ludzi?
Ostatnio w przestrzeni internetowej można natknąć się na informacje dotyczące jeleni zombie, a dokładnie przewlekłej choroby wyniszczającej (ang. chronic wasting disease, CWD). Choroba ta dotyczy głównie jeleni wirginijskich oraz łosi i jeleni kanadyjskich. Występuje ona głównie na terenach Ameryki Północnej [1] i straszy odbiorców popkulturowych treści, jakoby miałaby zawładnąć w niedługim czasie europejskimi zwierzętami, wkrótce potem całym światem dzikich zwierząt i w ostateczności dosięgnąć również nas – ludzi.
Na samym początku warto sobie uświadomić, czym w ogóle są choroby prionowe i jakie potencjalnie stanowią zagrożenie dla człowieka. Choroby prionowe to niezwykle groźne choroby mózgu, są one zakaźne, nieuleczalne i śmiertelne [2]. Powodem oraz głównym czynnikiem ich niezwykłej śmiertelności jest białko PrPC, które występuje w organizmie ssaków naturalnie. Przez wiele lat funkcje tego białka były owiane tajemnicą, jednakże dziś wiemy, że odgrywają one ważną rolę we wzroście aksonów oraz kierowaniu i różnicowaniu komórek glejowych w ośrodkowym układzie nerwowym (OUN) [3].
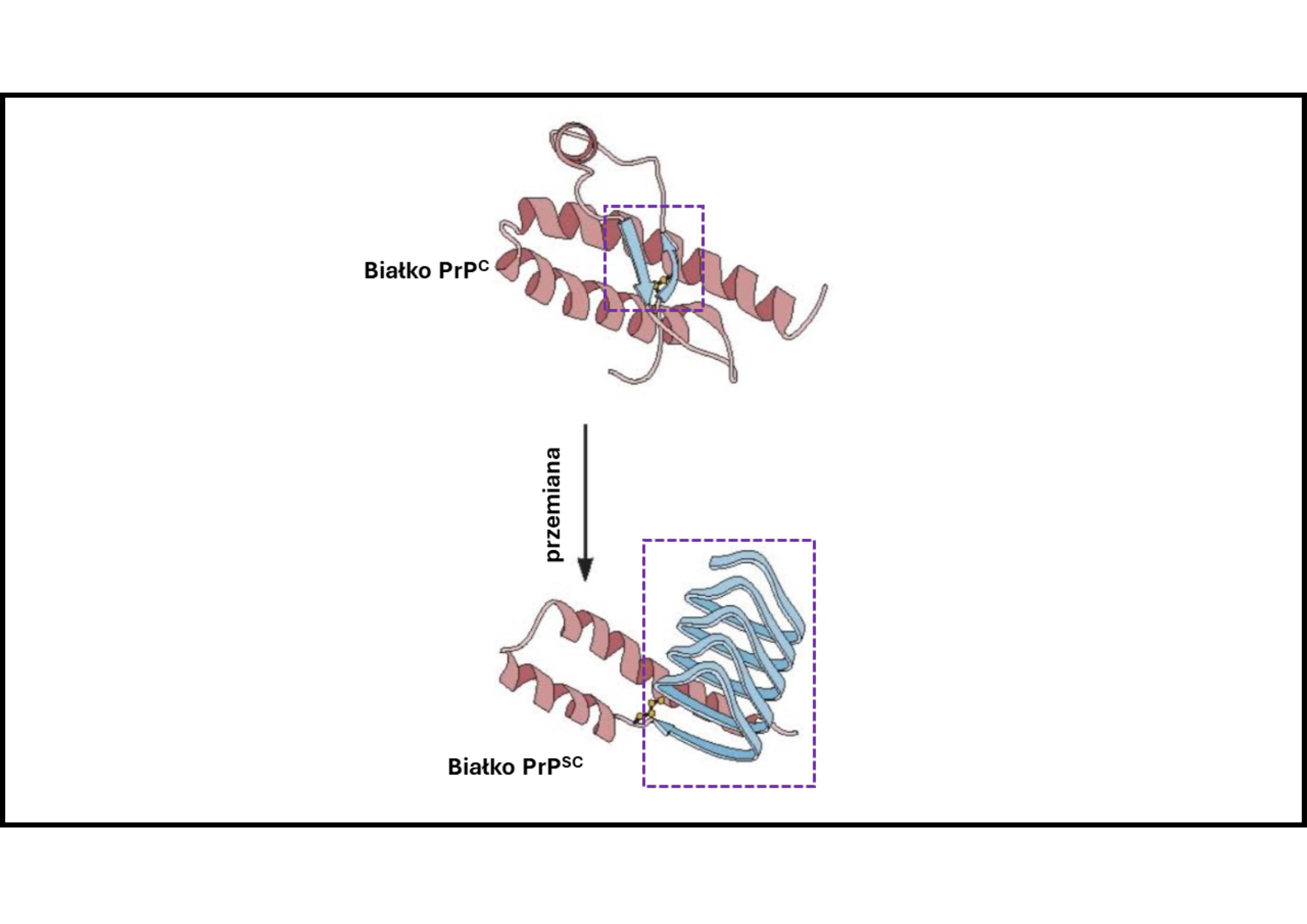
Rys. 1. Schemat przedstawiający zmianę strukturalną PrPc w PrPSC. Fioletowym kolorem zaznaczono zmiany struktury II-rzędowej.
Źródło: https://virology.ws/2016/09/15/structure-of-an-infectious-prion/
Jak to możliwe, że tak ważne białko jest powodem występowania neurodegeneracyjnych chorób?
Białko prionowe PrPC może przekształcić się w formę zakaźną PrPSC, która różni się od formy naturalnej zmianą struktury drugorzędowej białka – w przypadku formy zakaźnej występuje zwiększona ilość struktury β-harmonijki w porównaniu do normalnego wariantu białka PrPC, gdzie dominująca część dotyczy struktury α-helisy [4-9]. Dodatkowy problem stanowi odporność wariantu zakaźnego na działanie proteaz, co uniemożliwia zlikwidowanie czynnika chorobotwórczego przez chroniący się przed nim organizm [4,8].
Wstąpienie PrPSC może być samorzutne w wyniku nieprawidłowej ekspresji białka PrPC, wówczas powstałe białko PrPSC wpływa na zmianę zdrowych wariantów białka PrPc na drodze „reakcji łańcuchowej” zamieniając zdrowe warianty w warianty chorobotwórcze, kolejnym źródłem zakażenia jest dziedziczenie mutacji genu odpowiedzialnego za ekspresję wariantu PrPSC , zakaźne priony mogą być przekazywane również poprzez przeniesienie z zakażonego już organizmu [5,9].
Najczęściej spotykaną chorobą prionową u ludzi jest choroba Creutzfeldta-Jakoba (CJD) oraz jej różne odmiany takie jak śmiertelna bezsenność rodzinna (ang. fatal familial insomnia, FFI) i zespół Gerstmanna-Strausslera-Scheinkera [2,9]. Jedną z popularniejszych chorób neurodegeneracyjnych u ludzi jest choroba Kuru, zwana inaczej śmiejąca się śmierć [2].
Szansa na przeniesienie chorobotwórczego czynnika PrPSC z zakażonego organizmu zwierzęcia na człowieka wywołuje panikę na wieść o jeleniach zombie, które cierpią na chorobę prionową tj. gąbczastą encefalopatię (ang. transmissible spongiform encephalopathy, TSE). Zachorowanie na TSE powoduje gromadzenie się nieprawidłowych białek w mózgu, następnie w wyniku postępu choroby tkanka sprawia wrażenie „podziurawionej” i „gąbczastej” [10,11]. Tak naprawdę może minąć rok, a nawet lata, zanim u zakażonego zwierzęcia wystąpią objawy choroby prionowej [2]. Gdy dojdzie do aktywacji choroby powoduje ona wyniszczającą kaskadę u zakażonego zwierzęcia, m.in. zaburzenie funkcji poznawczych, apatię, drastyczną utratę wagi oraz zaburzenia równowagi [12].
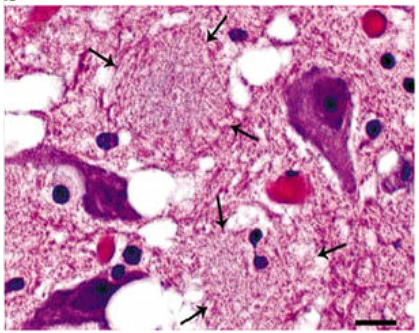
Rys. 2. Tkanka mózgowa z widocznymi „dziurami” powstałymi na wskutek choroby CWD [11].
Nie można wykluczyć przeniesienia choroby CWD na ludzi [12,13]. Badania in vitro dowiodły wystąpienie potencjalnego ryzyka zakażenia ludzkich komórek prionami CWD [14]. Na dodatek wykazano zakażenie małp, gryzoni i innych zwierząt, które miały kontakt z płynami ustrojowymi oraz tkankami zwierząt zakażonych CWD [11,12,13].
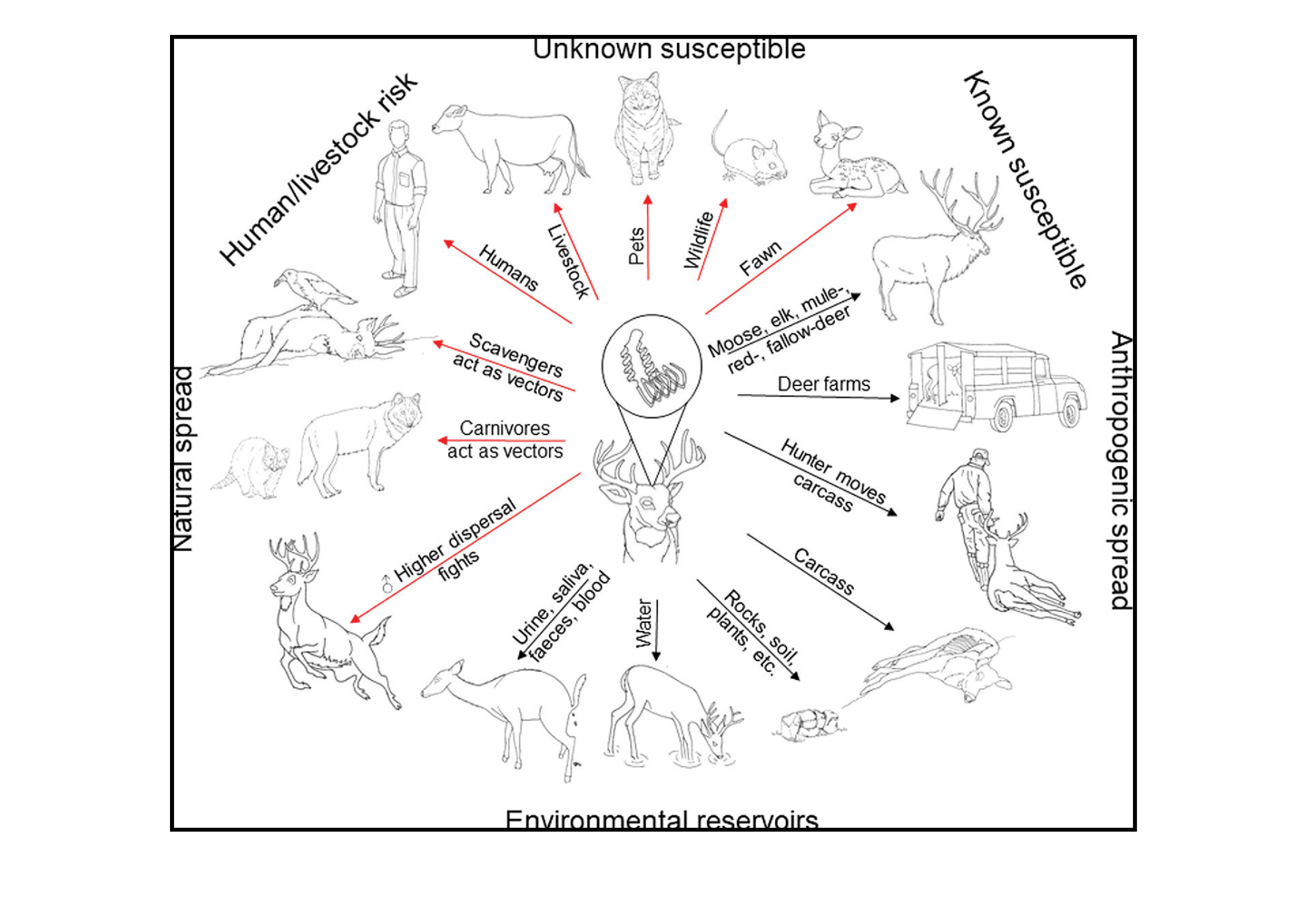
Rys. 3. Możliwe drogi rozprzestrzeniania się choroby CWD [13].
Na stan obecny chorobę CWD zaobserwowano w Kanadzie, USA, Korei Południowej, Norwegii, Finlandii oraz w Szwecji [1,13,15]. Wbrew pozorom nie jest to „świeżo” odkryta choroba, tak jak kreują to media, pierwsze przypadki CWD odnotowano w latach 60’ ubiegłego wieku na terenie USA, a w przypadku terenów Europy chore osobniki zaobserwowano w 2006 roku. Nie zmienia to jednak faktu, że choroba ta zbiera coraz to większe żniwo wśród zwierząt, stanowiąc również zagrożenie dla ludzi [1]. W Polsce dotychczas nie odnotowano ani jednego przypadku tej choroby, dodatkowo od 2018 roku wdrożono pierwsze działania mające uchronić polskie zwierzęta przed wyniszczającą chorobą CWD [1]. Warto pamiętać, aby spożywać dobrze przebadaną dziczyznę – oprócz ewentualnego zakażenia chorobą prionową mogą one posiadać również pasożyty, które zagrażają naszemu życiu.
________________________________________________________________
Literatura:
1) Flis M., Ścibior R., Przewlekła choroba wyniszczająca jeleniowatych (CWD) – działania prewencyjne, Życie Weterynaryjne, 2018, 93(4), 228-229.
2) https://www.niaid.nih.gov/diseases-conditions/prion-diseases (dostęp 03.02.2024)
3) Grimaldi, I., LeserF.S., Janeiro J.M. et al. The multiple functions of PrPC in physiological, cancer, and neurodegenerative contexts. J Mol Med., 2022, 100, 1405–1425, https://doi.org/10.1007/s00109-022-02245-9
4) https://www.scienceabc.com/pure-sciences/what-are-prions.html (dostęp 03.02.2024)
5) Kazula A., Kazula E., Choroby prionowe – charakterystyka, diagnostyka i terapia chorób prionowych, Farm Pol, 2009, 65(8): 594.
6) Birkmann E., Riesner D., Prion infection, Prion, 2008, 2(2), 67-72, DOI: 10.4161/pri.2.2.7060
7) Bartz J.C, Environmental and host factors that contribute to prion strain evolution, Acta Neuropathol, 2021, 142(1), 5-16. doi: 10.1007/s00401-021-02310-6.
8) Wille H., Requena J.R.,The Structure of PrPSc Prions,Pathogens. 2018. 7(1), 20.doi: 10.3390/pathogens7010020
9) Daniel G., Prion Protein Disease and Neuropathology of Prion Disease, Neuroimaging Clinics of North America, 2008, 18(1), 163-182,https://doi.org/10.1016/j.nic.2007.12.003.
10) Otero A., Duque Velasquez C., McKenzie D. et al. Emergence of CWD strains. Cell Tissue Res, 2023, 392, 135–148. https://doi.org/10.1007/s00441-022-03688-9
11) Gilch S., Chitoor N., Taguchi Y., Stuart M. Jewell J., Schatzl H. Chronic Wasting Disease. Topics in current chemistry, (2011), 305, 51-77. Doi:10.1007/128_2011_159.
12) https://www.cdc.gov/prions/cwd/index.html (dostęp 03.02.2024)
13) Escobar L.E., Pritzkow S., Winter S.N., Grear D.A., Kirchgessner M.S., Dominguez-Villegas E., Machado G., Townsend Peterson A., Soto C. The ecology of chronic wasting disease in wildlife. Biol Rev Camb Philos Soc. 2020, 95(2), 393-408. doi: 10.1111/brv.12568.
14) Barria MA, Libori A, Mitchell G, Head MW. Susceptibility of Human Prion Protein to Conversion by Chronic Wasting Disease Prions. Emerg Infect Dis. 2018, 24(8), 1482-1489. https://doi.org/10.3201/eid2408.161888
15) Rivera N.A., Brandt A.L., Novakofski J.E., Mateus-Pinilla N.E.. Chronic Wasting Disease In Cervids: Prevalence, Impact And Management Strategies. Vet Med (Auckl). 2019, 2(10), 123-139. doi: 10.2147/VMRR.S197404
